|
南方墨點法實驗流程
一、
限制酶切割
依據案件需求利用適當的限制酶進行切割
↓每個反應中含有下列成份:
DNA
16 μl
10X BSA
5 μl
10X Buffer
5 μl
Enzyme 30~50 U
補水至
50 μl
↓將上列混合均勻,放入適合限制酶切割溫度的培養箱反應隔夜。
↓確定是否反應完全,可先取少量進行電泳,若未反應完全,可在每反應添加限制酶30∼50
U,再繼續反應過夜後停止,進行電泳。
二、
洋菜膠電泳.
↓
製作 0.7
% 洋菜膠。
↓在經限制酶反應後的
DNA 溶液中添加
5 μl 10X dye。
↓混合均勻。
↓將
DNA 加入膠槽中,並加入1
kb marker
當標記,進行電泳。
↓先用
20 伏特
1小時進行電泳,再加高成
35伏特。
↓電泳時間視要分離的
DNA
長短而定。
三、
轉漬至尼龍膜
↓將電泳結束的膠照相,並利用投影片標記槽位及1
kb maker位置。
↓將膠體放入壓克力盆中並以二次水清洗
3 次。
↓加入適量
0.25N
HCl(要蓋過膠面),室溫輕微震盪
15 分鐘。
↓二次水清洗
3 次。
↓加入適量變性溶液【註一】(要蓋過膠面),室溫輕微震盪30分鐘。
↓二次水清洗3次。
↓加入適量中和溶液【註二】(要蓋過膠面),室溫輕微震盪30分鐘。
↓二次水清洗3次。
↓準備
3M 濾紙、耐龍膜(nylon
membrane)、吸水衛生紙。
↓將膠體背面朝上,進行南方轉漬如下圖。
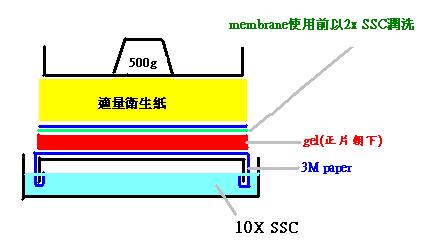
↓加入10
X SSC 約
2 L
。
↓吸水衛生紙需更換。
↓轉漬完成後,利用
2X SSC
清洗。
↓將
DNA 膜置於
3M 濾紙上,待微乾(表面無水漬)後放入UV-cross
linker 中進行
cross-linking(設定1200
x 100 μJ/cm2)。
↓將
DNA
膜包入鋁箔紙中,封住三邊留一邊(讓水蒸氣可以透出),置於80℃烘箱中,烘乾1小時。
↓取出保存於室溫。
↓進行雜交反應。
四、
雜交反應(Hybridization)
第一天:
↓預熱
pre-hybridization 溶液【註三】42
℃; 約
15 ml/DNA膜。
↓取適量
salmon
sperm DNA (ssDNA),100
℃水浴
8-10 分鐘,旋即置冰浴冷卻
2
分鐘(Working
concentration:100
μg/ml) 加入pre-hybridization
溶液,混合均勻。
↓桌上平鋪保鮮膜,取
mesh
鋪上,加約
50 ml 2X SSC潤濕。
↓取出
DNA 膜,使
DNA 面朝下,置於
mesh
上,將其潤濕後確定無氣泡後捲起,放雜交用玻璃瓶管中。
↓加入約
20 ml 2X SSC慢慢旋轉輕敲瓶子,使
DNA 膜與
mesh 慢慢展開緊貼於瓶內壁。
↓將
2X SSC
倒出,盡量倒乾。
↓將製備好的pre-hybridization溶液倒入。
↓放入42
℃雜交烘箱中,旋轉混均,孵育過夜。
第二天:↓預熱
hybridization 溶液【註四】42
℃約
30 ml/每瓶。
↓取適量
ssDNA,100℃
水浴 8-10
分鐘後旋即,冰浴冷卻
2
分鐘後加入hybridization溶液混合均勻
(Working conc.:100
μg/ml)。
↓
將
DNA 探針稀釋至
20~50 ng /45
ml TE (10 mM Tris-HCl, pH 8.0,1
mM EDTA)。
↓100 ℃水浴
5 分鐘旋即冰浴冷卻
2
分鐘。
↓將變性成單股之探針取45μl加入一管Radi-prime
(Amersham)中。
↓再取
5 μl α-32P
dCTP加入後混合均勻。
↓37 ℃,作用
30
分鐘。
↓加入
5 μl
0.2 M EDTA混合均勻以停止反應。
↓將標定好之探針利用層析柱〝Push
column〞【註五】純化。
↓將純化完成之探針於
100 ℃水浴
5 分鐘,旋即冰浴冷卻2分鐘。
↓將
42 ℃ 中的雜交瓶取出,將
pre-hybridization
溶液倒出 (盡量倒乾)。
↓將製備好之雜交溶液倒入,並加入適量製備好的探針混合均勻。
↓放入42
℃雜交孵育箱中,使旋轉混合過夜。
第三天:↓將42℃中的雜交瓶取出,將雜交溶液倒入貯存桶(含有放射性元素),將貯存桶置於壓克力箱中。
↓加入
50 ml solutionⅠ【註六】至雜交瓶後放入雜交烘箱中,旋轉混合數分鐘。
↓將solutionⅠ倒存於收集瓶,置收集瓶於壓克力箱中
(盡量倒乾)。
↓再加入solutionⅠ至雜交瓶中,放入雜交孵育箱中,旋轉混合數分鐘。
↓將solutionⅠ倒出(盡量倒乾)。
↓將
DNA
膜取出與
mesh 分開。
↓加入約
300 ml
solutionⅠ,振盪
20~30 分鐘。
↓使用蓋格計數器
(GM
counter) 偵測溶液及DNA膜,若計數>50
cpm,更換solutionⅠ。
↓用solution
I 將DNA膜放射性計數沖洗稀釋至約
20~50
cpm。
↓若訊號仍
> 50 cpm,可將洗液換成solution
II【註七】。
↓若訊號仍
> 50 cpm,可加高溫度清洗,每次加高
5℃。
↓清洗完後以
2X SSC沖洗。
↓將
DNA 膜置於
3M 濾紙上,使
DNA 面朝上,用保鮮膜包起。
↓壓X光片感光
(-70
oC)
至適當時間後沖洗取得結果。
【註一】Denaturing
solution:10
N NaOH 100 ml
NaCl
175.5 g175.5 g175.5 g
加水至
2 L
【註二】Neutralization
solution:1M
Tris (pH 8.0)、1.5M
NaCl
Tris
base 242.2 g
NaCl
75.5 g
濃HCl
84 ml
加水至
2 L
【註三】
prehybridization solution:
Formamide 200 ml
20X SSPE
100 ml
50X Denhardts 40 ml
10% SDS
4 ml
補水至
400 ml
【註四】hybridization
solution:
Formamide 200 ml
20X SSPE
100 ml
50X Denhardts 40 ml
50%Dextran 40 ml
10% SDS
4 ml
補水至
400 ml
【註五】
利用〝Push
column〞(Stratagene)
純化
32P-DNA
↓將Push
column加80
μl STE
(100 mM NaCl, 20 mM Tris-HCl, pH 7.5, 10 mM EDTA)
潤濕。
↓將約
55 μl 探針反應物加入
column 中。
↓過濾收集於
Eppend.
管中。
↓再加入
80 μl
STE buffer。
↓過濾過濾收集於同一Eppend.管中。
【註六】solutionⅠ:
2X SSC , 0.1%
SDS
【註七】solution
II:
0.1X SSC , 0.5%
SDS
 【註八】
20X SSC
: NaCl
876.5 g
補水至5 L,pH
= 7.0 【註八】
20X SSC
: NaCl
876.5 g
補水至5 L,pH
= 7.0
Sodium citrate 441 g
|